Lebanese International University – School of Arts and Sciences

Lebanese International University
School of Arts and Sciences
Department of Biological Sciences
BIOC400 - Biochemistry Ⅲ
Prepared and Presented by
Warda Abdulrahman
Supervised by
Prof. Hassan M. Khachfe
Spring Semester
2021 – 2022
Lebanon, Bekaa Campus, Khyara, www.liu.edu.lb
Structural analysis of the N-terminal of apolipoprotein B-100 by homology modeling between Apo B-17 and lipovitellin
Abstract
Background: Apolipoprotein B-100 (Apo B-100) is the major protein in LDL particles and also considered as the largest protein in the human body. Several techniques have been used for the aim of determining its whole structure such as X-ray and NMR-analysis but its large size impeded these attempts which required the use of other techniques for determination of each of its domains separately.
Result: Homology modeling is the technique followed in this paper and it involved the structural characterization of Apo B-100’s N-terminal based on the determination of the structure of Apo B-17 that forms the N-terminal domain of Apo B-100 using LV as a template structure due the fact that LV and Apo B-17 also perform the same activity of binding and transporting lipids.
Conclusion: Results obtained by electron microscopy of LDL particles containing Apo B-100 matches with the corresponding studied region of Apo B-17, this allowed the identification of the N-terminal of apo B-100’s structure.
Keywords: Apolipoprotein B-100, Apolipoprotein B-17, Lipovitellin, Homology modeling, LDL
Introduction
Atherosclerosis is a disease in which a plaque builds up inside the arteries, it is a severe illness and the predominant cause of cardiovascular diseases (CVD). Among the several factors that may initiate atherosclerosis, LDL particles also known as the bad cholesterol are the major causes. Apo B-100 is required for LDL, VLDL, and chylomicrons formation. The only protein component in LDL is a single molecule of apoB-100 per particle and it serves as a ligand for LDL receptors to bind and hydrolyze LDL. Consequently it is assumed to play a vital role in both the maintenance of cellular cholesterol homeostasis and the development of atherosclerosis. It was predicted that Apo-B is made of five domains in the order of NH3-𝜷𝝰1-𝜷1-𝝰2-𝜷2-𝝰3-COOH. Apo B-100 is a large glycosylated particle made of 4536 amino acids which has made the study of its overall structure at once quietly impossible. For that reason, it was suggested that the investigation and characterization of individual parts of the Apo B-100 particle separately would make it easier for the determination of its complete sequence and structure.
The N-terminal of Apo B-100 which is 𝜷𝝰1 comprises the first 1000 residues.
In addition to that, by electron microscopy, it was found that it is a globular structure and is made of alpha and beta secondary structures same as LV.
Actually, the N-terminus of Apo B-100 is made of an Apolipoprotein B-17 (Apo B-17) which contains six disulfide bonds and forms 17% of the full length of Apo B-100 sequence. In this article, we provide a homology-modeled framework for the Apo B-17 based on the crystal structure of lipovitellin (LV). LV and Apo B-17 are highly similar in structure and function where both of them are involved in binding to lipids and their transport. LV is a globular lipoprotein that is noticeably similar to the N-terminal of Apo B-100 (>30% similarity), and that is what makes it ideal as a template for the determination of Apo B-100 sequence and structure.

Figure1
The sequence alignment of B17 with the N-terminus of LV. The figure also shows the different structural domains (Cyan boxes are for the βC-sheet and the magenta boxes are for the α-domain. The underlined stretch indicates the region of no electron density in the template structure.
Results and Discussion
“The agent provocateur” was the name given to LDL as it was assumed to be the major cause of atherosclerosis. Based on the crystal structure of LV which is used as a reference for the structure of Apo B-17, we offer in this paper a detailed and a clear modeling approach for the structure of the N-terminal domain of apolipoprotein B-100.
𝜷𝝰1 domain of Apo B-100, is clearly homologous to LV, thus this domain and LV share similar structural features.
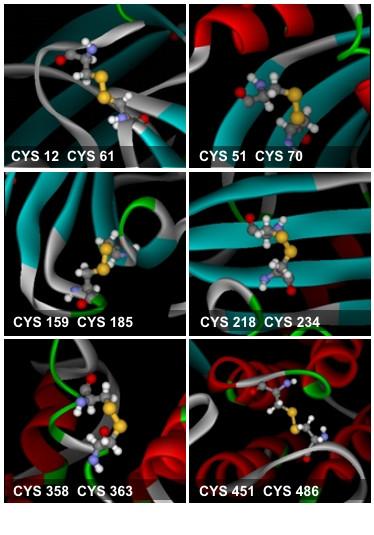
Figure 2
The six disulfide bonds found in B17. The cysteine residues are rendered in ball-and-stick representation. The sulfur atoms are colored in yellow.
Regarding the structure and constituents of LV, it is an egg yolk lipoprotein made of four 𝜷-sheets designated in sequence as 𝜷C-𝜷A-𝜷B and lastly 𝜷D. It also has a single 𝝰-helix located between 𝜷C and 𝜷A sheets. A lipid pocket is formed by the sheets B, D, and A in which lipids are transported through the body. Multiple sequence alignments demonstrated that the Apo B-17 sequence actually matches only with the C-sheet, the 𝝰-domain, and a portion of the A-sheet of LV.
Only one of the six disulfide bonds observed in Apo B-17 was retained in LV, therefore the remaining disulfide bonds that needed to be formed required precise investigations and tests to minimize steric hindrance. Since disulfide bonds are formed between sulfur atoms of cysteine residues, these atoms were moved to places where they were only separated by a distance of 4Å in order for bond formation to be possible. This was done by applying a set of automated or directed energy minimization steps only for residues that were more than 4Å apart (Figure 2).

Figure 3
The secondary structure prediction of the unstructured section.The secondary structure of this C-terminus of B17 was separately modeled, and the results from PredictProtein are shown, where the H's underneath the sequence indicate helical regions.
Since the region of the A sheet in LV had no correlate relation with the corresponding region in the Apo B-17 which is made of residues between 706 and 782, the structure of this stretch had to be determined in another way. A completely helical structure in this stretch has been reported by several methods including Chou-Fasman, the SPDBV modality of Deep View, and the PROF methods of PredictProtein (Figure 3). A stable structure had to be formed, and since as the rule implies, to become stable, a molecule had to lose some of its free energy; repeating steps of energy minimization and simulation were performed whereas its ends were stabilized in space at locations corresponding to the structure amino acids immediately preceding and
following the main sequence's starting and end residues, respectively. The structure of this portion was then pinned to the equivalent extremities in the LV-modeled B17 utilizing the LIGATE modality in HOMOLOGY (Figure 4).

Figure 4
The structure of B17 modeled after the N-terminus of LV. β-strands are colored blue, whereas α-helices are colored red. Arrows and labels indicate the beginning and end of the 78-aa stretch modeled independent of the LV structure.
This 𝝰-helical secondary structure forms a highly-organized architecture by the aid of some histidine residues bound to the helices where they induce additional intermolecular interactions once they become protonated by the means of reduced pH.The aqueous solvent accessibility of these residues was evaluated, and their protonation following pH drop was verified.
According to the observations made, LV has a concealed salt bridge in its structure made between arginine and glutamic acid residues at positions 547 and 574 respectively. This bridge has been found to connect the two 𝝰-helices found in the 𝝰-domain which adds to the stability of LV. A similar salt bridge has been detected in Apo B-17 after examination. It links lysine and glutamic acid residues at positions 530 and 557 respectively. Those residues are protected from aqueous solvents and by this, they increase the stability of Apo B17’s alpha domain (Table 1).

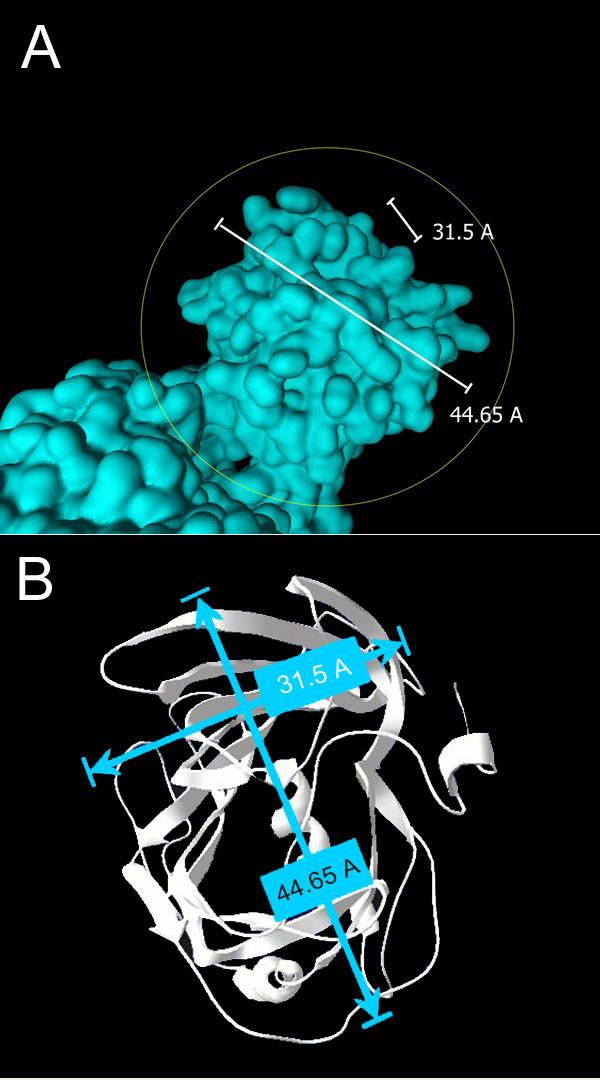
Figure 5
The spatial dimensions of the β-domain of B17. (A) Side view and (B) top view of the N-terminus of B17 with the corresponding dimensional values. These values correlate perfectly with the "knob" structure seen on the LDL particle and mapped to the N-terminus of B100.
Figure 5 shows the dimensions of the 𝜷-domain of Apo B-17 which are 31.5Å and 44.65Å. This confirms the similarity between the N-terminal of Apo B-100 and Apo B-17 since the same dimensions were found in a knob shape seen on the N-terminus of Apo B-100 when LDL particles were examined by electron microscopy.
Among the study of Apo B-17, sequence alignment was done using BLAST and a 60% similarity was detected between 26 amino acids in the C-terminal of Apo B-17 and a part of the constant domain of T-cell receptor. But since this paper aims to characterize the N-terminal of Apo B-100, we only focused on Apo B-17’s N-terminal.
The next step included the examination of the physical properties of Apo B-17, both bond angles and lengths were within the normal ranges of deviation from the mean standard and variability respectively. However, the RMS Z-score of these bonds had a percentage of approximately 9% away from that of LV. After this, distances between atoms forming bonds were studied and 23 out of a total of 31 bonds were found to have extremely short bonds so they were referred to be either hydrogen bonds or bonds between atom having a B-factor greater than 90 implying that the atoms presumably involved in these disturbances were not even present there.

Figure 6
Fold versus angle stability: P623 and T651, with Z-scores around -3.0, fall in turns involved in a mini sheet between the helical region and the C-terminus. The elevated torsion energy is compensated by an overall fold energy reduction.
Figure 6 shows a proline residue at position 623 of a 𝜷-strand and a threonine residue at position 651 which falls in 𝜷-turn joining 2 out of 3 strands forming a sheet between the helical region and the C-terminus of Apo B-17. Furthermore, The last figure (figure 7) shows that the Ramachandran plot of phi and psi angles of Apo B-17 and lipovitellin (LV) are practically identical.

The Ramachandran plots of B17 and LV. More than 97% of the backbone angle-pairs in B17 fall within the favorable regions, whereas 2.3% fall within the additionally allowed regions, and less than 1% is in the disallowed regions. This is very similar to the data from the LV system (98% in the favorable regions, 1% in the additionally allowed regions, and 1% in the disallowed regions).
Conclusion
As a conclusion, this study of the N-terminus of Apo B-100 is an important first step in the determination of the complete sequence and structure of this multi-domain particle. For further understanding of the role of Apo B-100, in vitro experiments and tests should be carried out, for example the direct imaging method offers a realistic tool for studying the structure of lipoprotein particles at the molecular level and thus helps achieving the main purpose of understanding lipoprotein conformation in vivo. Looking at the importance of Apo B-17 structure in the fully-completed structure of Apo B-100, this molecule will play a very important role in the synthesis of drugs that will be efficient in the treatment of patients with cardiovascular diseases or any other ones caused by increased concentration of LDL in the blood. These drugs may be also useful in ensuring protection for patients having risk factors of developing CVD.
Methods
Molecular modeling
Sequence alignment
Multiple sequence alignments were done using BLAST [32] and the alignment module of the Discovery Studio suite (Accelrys Inc., Discovery Studio 1.5, San Diego:
Accelrys Inc., 2004)
Structure prediction
The structure of B17 (residues 1–704) was modeled using MODELLER [33] of HOMOLOGY in insight II (Accelrys Inc., Insight Modeling Environment, Release 2000.1, SanDiego: Accelrys Inc., 2002), based on the crystal structure of lipovitellin (LV), an egg yolk protein that shares over 30% sequence homology (in over 700 amino acid overlap) with B17. The secondary structure of the unstructured region was predicted using the Chou-Fasman Algorithm [26], the PROF methods [27,28] and the Deep View modality [34]. The calculation was performed using the Accelrys SeqWeb server of the GCG Wisconsin Package.
Simulation
Energy calculations
EC's were performed using DISCOVER (Accelrys Inc.,CDiscover Molecular Simulator, Release 2000.1, SanDiego: Accelrys Inc., 2002) and CHARMm (Version c28b)
[35] modules in Insight II. Energy minimizations were performed using the Steepest Descent method followed
by Conjugate Gradients.
Molecular dynamics
MD Simulations were carried out with periodic boundary
conditions using a cubic box (of appropriate size), in the Insight II package. Solvent water molecules were represented by the three-site TIP3P water model [36], in the NVT ensemble.
Force fields
Calculations were performed using the DISCOVER force-fields CVFF and CFF91. The CHARMm force-field used in the solvation simulation was CHARMm27.
Analysis
Solvent accessibility
Solvent Accessible Surface Area (SASA) was calculated for individual atoms using the Structural Biology at NIH server (Structools), with a probe radius of 1.4 Å [37].
Potential maps
Solvation energy and hydrophobic interactions were calculated using the Delphi module in Insight II (Accelrys Inc., Delphi Module, Release 2000.1, San Diego: AccelrysInc., 2002), using the CFF91 force-field. Potential maps were constructed using a grid. The dielectric value was assigned as 4 for the protein and 80 for the solvent.
Structure validation
Structure validation tests were carried out using the PRO-CHECK [38] and WHATIF [39] modalities. Calculated values were referenced to the reported mean standard values[31].
References
Copied from “The N-terminal domain of apolipoprotein B-100: structural
characterization by homology modeling” article written by Hassan Al-Ali and Hassan M Khachfe and published in BMC Biochemistry journal on 22 July 2007.
.png)

.png)
Your project is very interested I like the clarity and sequence of your ideas add to the effective and ordered images and graphs. Keep going wardda you are the best.
ReplyDelete